 By Jeff Elton, PhD, CEO
By Jeff Elton, PhD, CEO
In 2021, the generation of evidence in support of regulatory decisions changed forever and fundamentally. This shift was not sudden or random. Rather, it was the result of years of work, debate, research, pilots, and deliberate decisions. It was the year when advanced retrospective analyses; retrospective data integrated into prospective studies; productive studies designed with retrospective data; Artificial Intelligence (AI) technologies to identify patients for study eligibility; and digital-only prospective approaches all emerged and created the foundation for something new – essentially an “Evidence Generation SystemTM” that can operate over the entire life cycle of a therapeutic program, or even a disease.
Many factors signaled this exciting development: the confluence of the FDA seeking comment for new Real-world Data (RWD) guidance; use of RWD in COVID-19 studies; accelerated capture of data from more settings; increased use of technologies for linking data across sources; adoption of RWD deeply across all aspects of clinical development planning and operations; and RWD producing evidence for new diagnostic, therapeutic and vaccine approvals.
The Health Information Technology for Economic and Clinical Health Act (HITECH), which took effect in 2010, required electronic medical records (EMRs) in American healthcare to comply with standards set by the U.S. Department of Human Services’ Office of the National Coordinator for Health Information Technology. This mandate essentially created a common data foundation for the entire country – something unique to the US. It was always anticipated that this data foundation would enable more healthcare research. The 2010 Patient Protection and Affordable Care Act further catalyzed innovation with the creation and foundation funding of the Patient-Centered Outcomes Research Institute as a US-based non-profit organization championing pragmatic trials. Pragmatic trials are designed to evaluate the effectiveness of interventions in real-life routine practice conditions, often using EMR data as the sole source or a major source of data.
Broad access to clinical data in electronic formats gave rise to two additional areas of work that helped create the foundations of RWD and more robust study designs to generate evidence: natural language processing (NLP) and unstructured data abstraction supported by advanced Software-as-a-Service (SaaS) solutions used by clinical experts.
NLP had its foundations during the intelligence and translation activities of the Second World War. After the war, there was extensive work to translate one language into another while retaining grammar, structure, and meaning. Noam Chomsky’s work from the 1950s through the early 1970s aligned linguistics and computer science and defined expectations that machine processing of words and concepts should be comparable to that of a native language speaker. These developments drove most research and applications until stochastic researchers integrated optical character recognition and pattern recognition to the reading and comparison of texts. As computation power progressed and was available to more researchers, logic-based paradigms emerged that demonstrated how machines could both ‘understand’ and ‘learn.’ Terry Winograd’s work during this period showed these advancements in limited domains. By the late 1980s and through the 1990s, empirical, probabilistic, and statistical models emerged, allowing machine ‘reading’ to include context and the likelihood of meaning. With vast amounts of health data emerging in both medical claims and EMRs, the foundations were in place for processing vast amounts of information at scale and speed as determined by the number of allocated cores of computation power.
In parallel with NLP advances, unstructured data curation with specialized software tools and sets of concepts, or ‘rules’ for consistently deriving machine-readable data or variables, have also evolved. Here, too, is a long-term history of medical coding that goes back to the late 1500s in the different parishes that would ultimately constitute the City of London. By 1611, the Worshipful Company of Parish Clerks was formally arraying the sources for deaths with cause. These highly manual systems, which involved transcribing data from one paper record system into another, persisted for hundreds of years until the modern EMR systems appeared in 1972 at the Regenstrief Institute. Today, local healthcare professionals at the institution generating the clinical data generally perform data coding and unstructured data abstraction in the belief that they will know the data, diagnostic processes, documentation protocols, and treatment regimens of their respective institutions more than others. Also, it has been assumed that local clinical personnel are in the best position to navigate personal health information found in unstructured data in manners appropriate for specific research questions and quality-of-care analyses.
There are also new specialized companies, operating under business associate agreements, with large clinical workforces trained in specific rules, for specific clinical concepts, with data source codifications, and consistently applied source-specific rules. The advantage of these approaches that sit above any one healthcare provider is the consistency of the rules and therefore of the meaning of a concept. ConcertAI itself is a leader in both advanced NLP and software-enabled, clinical-expert-executed curation. Here the choice of approach is often the intended use-case where broad epidemiological studies benefit from great scale and NLP versus narrow questions that are presented to a peer review journal or in support of evidence generation for regulatory decisions.
As we look forward, we are poised for an integration of these two approaches. Already we are seeing the accuracy and recall of NLP models rivalling that of expert human data abstraction. The field of neural NLP is aiding even further advances. These models and solutions are beginning to augment the capabilities of expert humans across complex document formats, multiple EMR systems, and treatment patterns that might involve 200 or more specific concepts to develop, differentiate and track longitudinally. As data use and utility continue to increase, we can expect the integration of machine and human approaches to become standard. In fact, I would be surprised if machine ‘reading’ does not complement 100 percent of human-based data abstractions for parallel reads and quality control even as NLP and intelligence machine pre-processing will improve the accuracy and productivity of human abstraction. The recent FDA guidance documents for commentary may be seen as the formal beginning of that process. To quote from the September 2021 FDA document Real-World Data: Assessing Electronic Health Records and Medical Claims Data to Support Regulatory Decision-Making for Drug and Biological Products: “Technological advances in the field of artificial intelligence (AI) may permit more rapid processing of unstructured electronic health care data. Advances include natural language processing, machine learning, and particularly deep learning to: (1) extract data elements from unstructured text in addition to structured fields in EHRs; (2) develop computer algorithms that identify outcomes; or (3) evaluate images or laboratory results.”
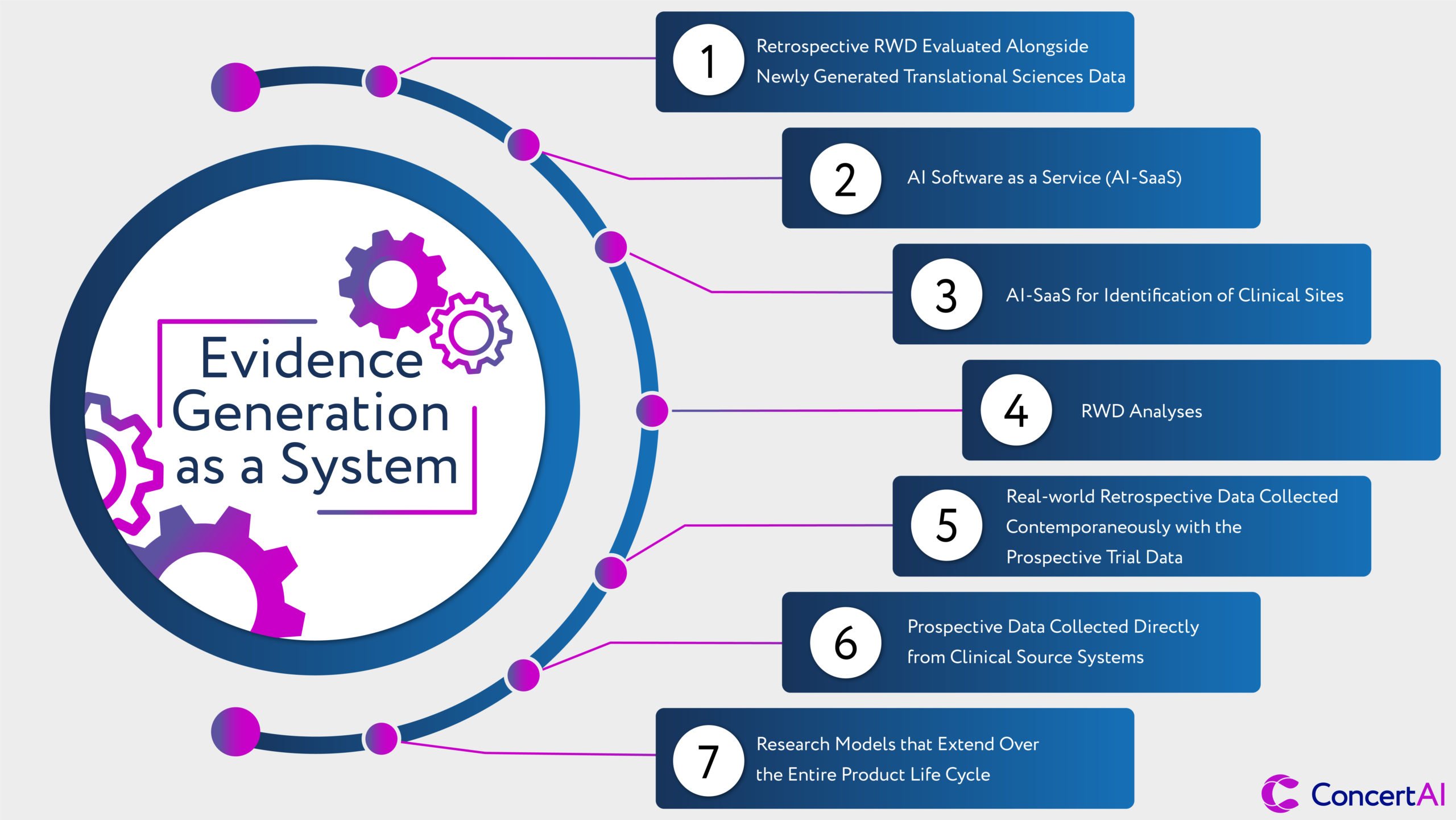
We have arrived at the foundation of a new evidence or research system. In this new system, different data sources, technologies, and study designs can be reconsidered to derive evidence of high reliability, accuracy, and representativeness. These new approaches will involve:
- Retrospective RWD evaluated alongside newly generated translational sciences data to consider early human studies that both provide a validation of new biological mechanisms or the value of new therapeutic entities for patients relative to the current standard(s) of care.
- AI Software as a Service (AI-SaaS) solutions that can optimize clinical study designs for broad representativeness, utility for regulatory decisions, and avoidance of legacy biases in study access.
- AI-SaaS for identification of clinical sites for potential participation in a clinical study and for the ultimate identification of patients within clinical workflows of the participating healthcare providers. This process will use AI models and algorithms to ‘read’ patient records and integrate EMR, molecular, and imaging data together.
- RWD analyses as part of the initial consultative sessions with regulatory groups to provide a clearer view of the study intent; why it would be generalizable to the majority of patients who may be ultimately treated with the proposed therapeutic; and the incremental value to patients and the health system being targeted. Today this is largely Electronic Medical Record (EMR) data, but in the near term this will include confederated medical claims data (both open and closed forms), and images from Picture Archiving and Communication Systems (PACS).
- Real-world retrospective data collected contemporaneously with the prospective trial data as part of a further standard-of-care validation of prospective benefits, or perhaps as a replacement for traditional randomized control trial controls – especially for rare diseases and where the standard care is entirely inadequate. The power of ‘matched’ data – where the prospective and retrospective data come from the same patient populations and treatment centers – is enormous and enhances confidence in the generalizability of results for the entire population that may have access to a newly approved medicine.
- Prospective data collected directly from clinical source systems will follow an ‘enter once and use many times’ approach to research. As the original intent of the HITECH act’s consistent implementation of electronic medical records, this shift holds the promise of clinical research being no more complex for patients or providers than the standard of care itself while accelerating needed medical innovations and allowing broader study access to patients. These data will be derived from EMR, Laboratory Information Management Systems (LIS), and PACS for radiologically acquired image series.
- Research models that extend over the entire product life cycle, beginning at the point of completion of registrational studies, and with approvals to provide ongoing insights in treatment durability, long-term efficacy, and long-term safety. These new research approaches would be continuous with the original registrational studies and any requirements that may be part of an approval.
These are the elements of the distinctively performing and leading research organization of the future. A future that integrates multiple data types and sources; that brings together the best approaches of data sciences and biostatistical analysis; that sees research approaches as optimized for results, speed and efficiency; and finally, one that assures broader access and broader applicability of the research results to the 80% of patients treated in the community. We have the data, technologies, provider networks, and regulatory receptivity to make bolder strides. In the end, these changes will enable us to conduct better research, faster research, and more research.
ConcertAI is dedicated to creating the “evidence armamentarium” for oncology and hematology. We take pride in our independence, lack of conflicts, diversity of data sources, and tri-part commitment to patients, providers, and biomedical innovation. Already we present the largest collection of research-grade data sets across major solid tumors and hematological malignancies, including some designed for pan-tumor targets of treatment approaches. We are now advancing unique solutions that integrate AI models, software, and RWD for study design as a single solution – neither a standalone data set nor a SaaS solution but rather a purposeful integration of both. The same healthcare providers where we partner for retrospective research are now being powered with patient identification tools for prospective clinical studies. And these same clinical centers are now being enabled with leading prospective research solutions that acquire data from clinical source systems – EMRs, PACS, LIS, etc. – for registrational and post-approval studies.
We can now define critical clinical questions impacting the majority of patients, design clinical trials informed by these insights, bring together retrospective data for consultative regulatory conversations, and run prospective studies with retrospective elements from the same broad array of clinical sites and data sources that are complementary and internally consistent. These capabilities allow for the most robust analyses, the highest standards of reliability, greater source-data provenance and increasing confidence in the generalizability of study outcomes to the majority of patients. This is what biomedical research is supposed to be. This was the basis for the original 21st Century Cures Act’s mandate for RWD, and this is why the FDA’s most recent guidance was formalized and presented for comment and finalization this last September and November.
As we look to 2022 and beyond, we could not be more enthusiastic for what lies ahead and the benefits to be created from this emerging “System of EvidenceTM” for accelerated biomedical innovations, new AI-augmented technologies for providers, and patients with the highest unmet needs.

